K computer Helps Solve Mystery of Bacterial Drug Resistance
Explaining living phenomena in physical terms
K computer Helps Solve Mystery of Bacterial Drug Resistance
Explaining living phenomena in physical terms

Research Scientist with the Computational Biophysics Research Team, RIKEN AICS
One surprising finding Matsunaga discovered during his research of AcrB was that it took far more time to analyze the simulation data than to carry out the actual simulations using the K computer. This has led him to seek out the help of machine learning and artificial intelligence to help speed up this process.
Pathogenic bacteria that have become resistant to medical drugs pose a serious threat to clinical practice. In such cases, the drugs are driven out of the body by the multidrug efflux transporter AcrB protein embedded in the bacteria’s cellular membrane and become almost ineffective. To work out the mechanism causing the efflux or discharge of a drug, Matsunaga simulated AcrB’s functions on the K computer. He discovered that drug efflux takes place as the protein undergoes a transformation when binding with just one hydrogen ion.
When Matsunaga enrolled in the Department of Earth and Planetary Sciences (EPS), Faculty of Science, Kobe University, in 1997, he spent much of his early campus life reading extensively. During this period, he came across a book that would impact the rest of his career. The title was What is Chaos? by Keisuke Ito, who was a professor in EPS at the time.
Ito’s book triggered Matsunaga’s interest in Chaos Theory—the phenomenon of how small changes in a system can later lead to unexpected outcomes. “I became drawn to computer simulations of chaos because it was fun to see simulations produce unpredictable results one after another,” he recalls. In obtaining his Master’s degree working under Professor Yukio Gunji and Associate Professor Tamiki Komatsuzaki at EPS, he took up the study of simulation as a research tool in a serious way.
“Calculations using Newton’s equation of motion, F (force) = m (mass) x α (acceleration), makes possible a variety of simulations, from the orbits of planets to the movements of proteins,” says Matsunaga. “Even in simple simulations, like those of movements of planets in our solar-system, chaos could produce surprising results. As a young researcher, I became infatuated with the charm of Newton’s F=mα, and set myself the goal of mastering the secrets of the equation.”
After earning a D. Sc. in information and media science, Matsunaga joined the lab of Professor Akinori Kidera in Yokohama City University in 2007 and was later assigned to research AcrB using simulation.
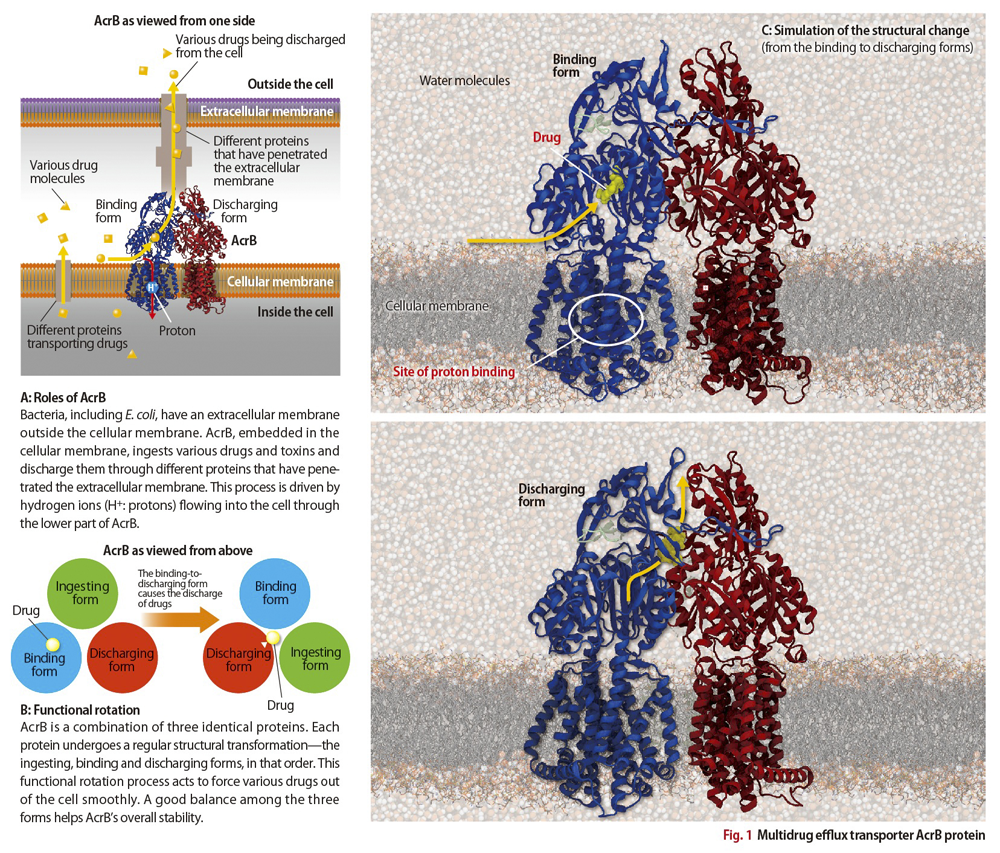
AcrB is a protein embedded in bacterial cellular membranes (Fig.1A). As molecules of various drugs enter the cells of bacteria, the protein ingests then discharges them, and in this way the bacteria develop a resistance to the drugs.
When Matsunaga began investigating AcrB, previous researches had already produced a partial picture of the protein’s shape and the discharge mechanism.
Most proteins, including AcrB, have a three-dimensional structure composed of a folded amino-acid chain. Never static within a cell, a protein morphs into various structures to perform its functions. These changes can be simulated using calculations based on interatomic-forces and the F=mα equation. Vital to such simulations is information obtained at the atomic scale on the shape of the protein and on the kind and location of the atoms. The most common way to obtain such data is X-ray crystallography, the technique of applying X-rays to protein crystals to analyze their structures.
Professor Satoshi Murakami, currently at the Tokyo Institute of Technology, was the first to reap major results from the X-ray crystallography approach. In a 2002 analysis using RIKEN’s SPring-8 synchrotron radiation facility, he revealed AcrB’s structure and showed it to be a combination of three identical proteins.
In 2006, Professor Murakami followed this revelation with a successful experiment, also conducted on the SPring-8, that examined the structure of AcrB when a drug molecule is bound. He discovered that each of the three proteins mutually differs slightly in structure, and that each structure corresponded respectively to the forms that ingested, bound, and discharged the drug molecules.
Professor Murakami surmised that each AcrB protein changes its structure in sequential order to perform these three actions (Fig.1B). Then, what drives this sequential change?
It is known that E. coli and other bacteria have an extracellular membrane. And between it and the cellular membrane protons (hydrogen ions) are present in high concentrations. It is also known that the protons flow into bacterial cells through the membrane proteins. Thus, it seemed likely that AcrB’s structural change takes place as the protons permeate through the protein’s lower region immersed in the cellular membrane (Fig. 1A).
In other research, Professor Mitsunori Ikeguchi at Yokohama City University had identified the binding site of protons that would mainly affect AcrB’s structure. It remained to be seen, however, what the exact structural change was that caused the discharge of drugs. Matsunaga was given the task of answering this question using computer simulation.
“AcrB is a complex organic compound, consisting of as many as 500,000 atoms including those of the surrounding water molecules and cellular membranes,” Matsunaga explains. “Simulating its activity by evaluating interatomic forces and repeating F=mα calculations entails an enormous amount of calculation.”
After joining AICS in 2011, Matsunaga sought to devise effective calculation methods to simulate AcrB on the K computer. His efforts were eventually rewarded. He succeeded in reproducing the process of structural change in AcrB when the proteins bind with the protons—the process that results in a discharge of drug molecules (Fig.1C).
An animated illustration is available here.
AcrB proteins ingest and discharge drugs in their upper region, which is relatively far from the lower region where the protons permeate. “It’s hard to imagine that a single, large ‘system’ of 500,000 atoms would undergo a structural change—from a binding structure to a structure that discharges foreign molecules—merely upon the adhesion of just one proton,” says Matsunaga. “Figuratively speaking, it’s like an elephant being irritated by an ant walking over its foot.”
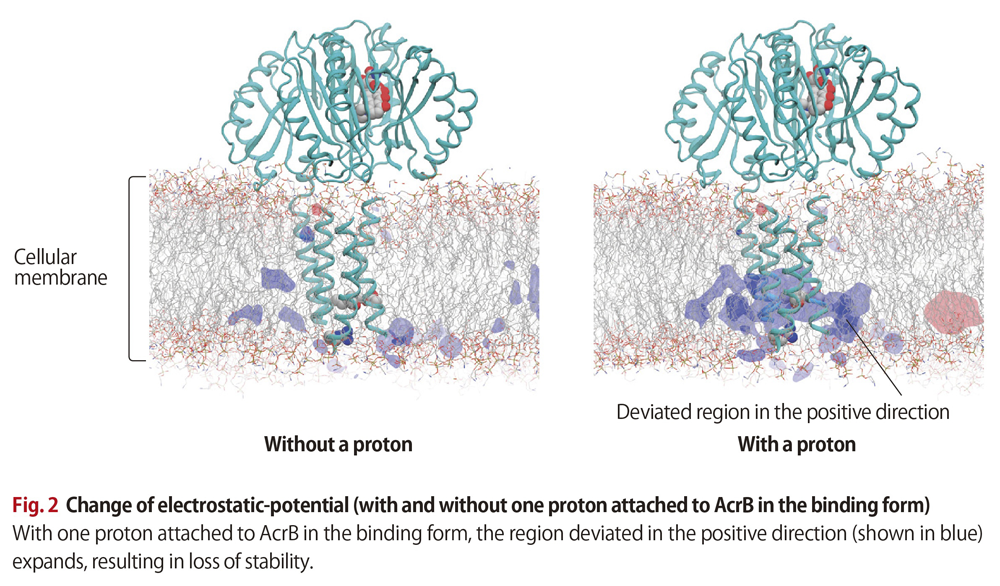
Matsunaga’s simulation work produced some surprising findings. When AcrB proteins in binding form are without protons, practically no electrostatic-potential frustration appears in either the positive or minus direction. Yet, when coupled with just one proton, the proteins experienced a major expansion of the region that deviates in the positive direction. This resulted in the proteins losing much of their electrostatic stability. “A large stability loss from just one attached proton!,” Matsunaga exclaims (Fig. 2) .
The simulations also showed AcrB’s lower region changing structurally, as if to alleviate the frustration. This, in turn, affected the upper region and resulted in the structural change that discharged the drug.
Previous research showed that some specific amino acids in the protein’s lower region also play a role in this structural change. “Given the amount of stability loss, there must be other important factors involved as well,” says Matsunaga. “I am looking for these by comparing the two states of AcrB in the binding form: one with the single proton, the other without.”
An inhibitor drug is any molecule that binds and deactivates a protein ultimately responsible for disease. As for AcrB, its drug-discharge functions may be inhibited by deactivating not just the drug-binding upper region but also the site that informs the upper region of a change in the proton-attached lower region.
Thus a better understanding of a protein’s functional mechanism should help expand the number of options when targeting sites with inhibitor drugs. It is expected that the strategy of targeting other sites than the obvious binding sites will lead to the production of new drugs for proteins previously hard to deactivate.
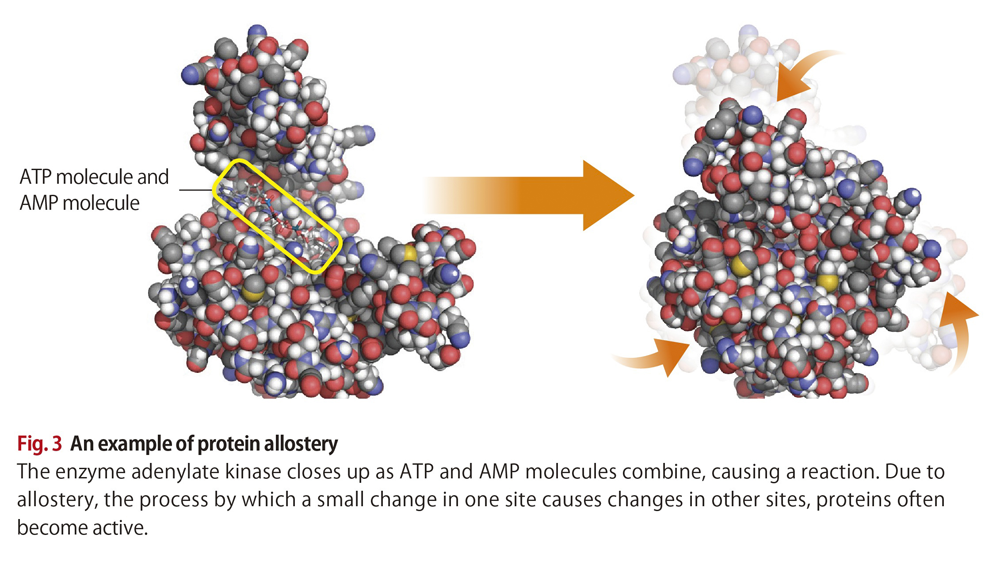
The way AcrB functions represents an interesting example of allostery. Allostery refers to the process by which a small change at one site causes a change in a separate site.
Thanks to allostery, a range of proteins are able to carry out their functions (Fig. 3). “There should be a hidden thread running through all this, and I would like to determine what it is by way of simulation work,” says Matsunaga. “To this end, I’m looking to see how machine-learning technology could draw out common features in simulation data that we humans can miss. These features will most likely assist development of new drugs.”
An animated illustration is available here.
Calculating the activity of proteins, each consisting of a very large number of atoms, requires an enormous amount of mathematical work and computer processing time. As such, researches’ ability to simulate protein activity has been limited to mere microseconds (millionths of a second). Matsunaga, however, using a new method of calculation together with the power of the K computer has succeeded in extending the amount of simulation time a thousand fold—on the order of milliseconds (thousandths of a second).
“This now makes it possible to directly compare the results of simulations and experiments,” he says. “That will lead to a clearer understanding of life phenomena. Once we predict, through simulation, that a particular site in a protein likely has a lot to do with the protein’s overall functions, we should be able to verify the prediction by experiments that modify the site and examine the functional changes brought about by those modifications.”
Though experimentation will enable researchers to compare protein conditions before a structural change with conditions after the change, it is difficult to observe these changes in real time. The same is true of AcrB.
“AcrB in the binding form, with one proton attached, enters an unstable state for an extremely brief period, too brief for observation,” he points out. “But, using simulation, we are able to examine the key moments of protein functions at the atomic scale and explain life phenomena in physical terms.”
Life science is now at the threshold of another breakthrough, where computer simulation serves as a primary tool of research together with experiment.