


- メニュー
創薬とスパコン

新しい薬を生み出すには化学合成や動物での実験が必要です。でも、その前にスパコンの出番があります。薬の設計にスパコンがどのように役立つのか、ポスト「京」では何が計画されているのか、ご紹介しましょう。
関連する課題:重点課題(1)生体分子システムの機能制御による革新的創薬基盤の構築
薬はどのようにつくられるのか
薬はなぜ効くのか、知っていますか? 病気になったとき、私たちの体の中では、病気の原因になる分子(おもにタンパク質)が活発に働いています。薬は、病気の原因分子に結合し、その働きを抑えてくれるのです。例えば、胃潰瘍の薬であるシメチジンは、胃酸の分泌をうながすタンパク質の働きを抑えます。だから、胃酸の分泌が抑えられ、胃のむかつきやもたれが治るのです。
もう少し詳しくいうと、このタンパク質はヒスタミン受容体というものです。このタンパク質には、体内にあるヒスタミンという化合物がぴったり入るくぼみがあります。これは、鍵と鍵穴の関係にたとえることができます。そして、ヒスタミンという鍵が受容体の鍵穴に入ると、この受容体が胃酸の放出をうながすのです。これは私たちが生きていく上で必要な作用ですが、ヒスタミンがどんどんやってくると、胃酸が出すぎてしまいます。それを防ぐのが、シメチジンという薬です。シメチジンは、ヒスタミンとよく似た形をしていて、ヒスタミンが来る前に鍵穴に入るのです。そうなると、ヒスタミンは鍵穴に入ることができず、胃酸の分泌が抑えられるというわけです。
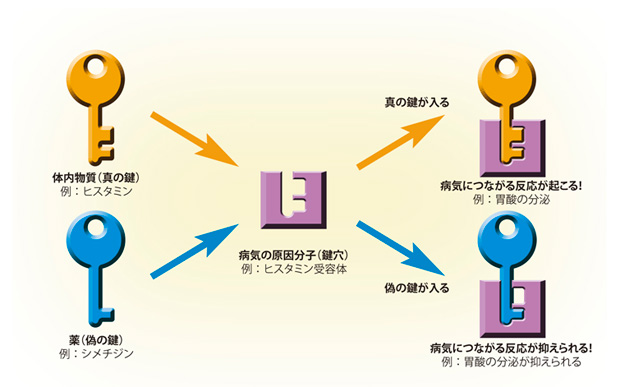
鍵と鍵穴
薬がなぜ効くのかは、病気の原因分子を「鍵穴」、そこに入る化合物を「鍵」にたとえるとわかりやすい。
シメチジンは1975年にイギリスで開発された薬で、「鍵穴をふさぐ」という方針でつくられた薬として先駆的なものです。それ以前につくられた薬も、実は鍵穴をふさぐものが多いのですが、それを狙ってつくられたわけではありませんでした。現在では、ほとんどの薬が「鍵穴をふさぐ」という方針で設計されています。まず、病気の原因となる分子を明らかにし(①ターゲットの決定)、それと結合する化合物を探し(②ヒット化合物の探索)、結合するものが見つかったら、その構造を少しずつ変えて、薬効、安定性、安全性などの面でよりよいものにしていく(③リード化合物の最適化)のです。
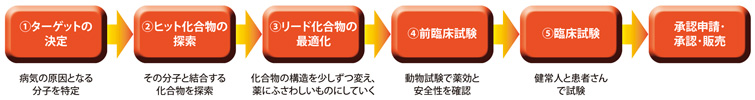
薬の開発プロセス
リード化合物が最適化されたあと、動物やヒトでの試験が行われる。1つの薬を開発するのに、11~15年の歳月と、1000億円程度の費用がかかると言われる。
従来、薬の設計は細胞や動物などを使った実験によって行われてきましたが、1970年代に、コンピュータの中で分子の形や結合を計算する方法が進歩し、1980年代以降、それらを薬の設計に使うための研究がさかんになりました。「鍵穴をふさぐ」という薬の設計方針には、計算が貢献できる部分が大きく、1990年代には、実際に、計算の力を借りて開発された薬が上市されるようになりました。初期の例として、緑内障治療薬「ドルゾラミド」(1994年にアメリカで認可)、インフルエンザ治療薬「ザナミビル」(1999年にアメリカで認可)などが知られています。
2003年にヒトゲノムが解読されると、ゲノムに基づいて病気の原因分子が特定されるようになり、また、原因分子の形も調べやすくなりました。現在では、計算は、薬の開発にとって欠かせない要素技術となっています。
鍵穴をふさぐ化合物を計算で探す
現在、製薬現場で計算がもっとも活躍しているのは、原因分子と結合する化合物を探す(上の図の②ヒット化合物の探索)段階です。
もちろん、実験で化合物を探す方法も使われています。非常に多くの種類の化合物を集めておき(これを「化合物ライブラリー」と呼びます)、原因分子と化合物を次々に混ぜて、結合するかどうかを調べるのです。この実験には手間がかかるので、1990年代から、専用の自動化装置を用いるハイスループット・スクリーニングという方法が使われるようになりました。

ハイスループット・スクリーニングのロボット(次世代がん研究シーズ戦略的育成プログラムのホームページより)
分注装置を備えており、一度にたくさんの化合物を原因分子と反応させることができる。
この操作を計算で行うのが、バーチャル・スクリーニングです。コンピュータの中に、原因分子の鍵穴の形をつくっておき、いろいろな化合物がそこに入るかどうかを調べます。原因分子のくぼみに化合物が入ることを「ドッキング」とよびます。
コンピュータの中でドッキングを行わせるための最初のソフトウェアは、アメリカのクンツらが1982年に開発したDOCKです。クンツらは、その当時、すでに3次元構造が実験でわかっていたミオグロビンというタンパク質を対象として、くぼみに化合物がどのように入るかを計算で明らかにしました。このとき使われたコンピュータは、ディジタル・イクイップメント・コーポレーション(DEC)のPDP-11/70 という16ビットのミニコンピュータ(当時の大型コンピュータより小型で安価なコンピュータ)で、計算時間は数時間だったそうです。
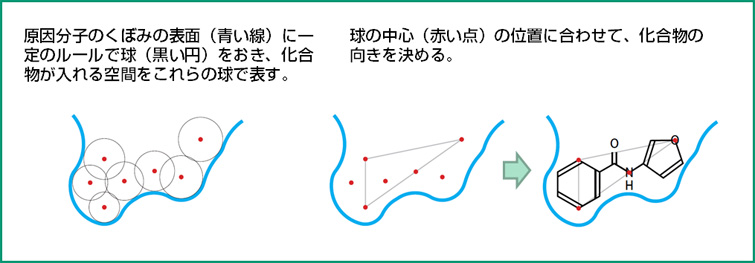
DOCKで化合物の入り方を調べる方法の原理
くぼみの形や、化合物の化学式はイメージ。

PDP-11/70(写真提供:Joe Mabel)
PDP-11/70のシリーズは、プログラムを書きやすいことから広く使われた。計算性能は「京」の1億分の1程度だったといわれている。
DOCKの誕生後、数多くのドッキングソフトが開発され、コンピュータの進歩とともに改良されてきました。現在では、製薬企業も、ドッキングソフトを利用して、バーチャル・スクリーニングを行っています。
バーチャル・スクリーニングが可能になった背景には、3つの要因があります。1つは、タンパク質の3次元構造を明らかにする技術(X線構造解析など)が進歩して、立体構造の知られているタンパク質がどんどん増えていったこと。2つめは、薬の候補となる化合物(形の情報を含む)を集めたデータベースが整備されたこと。そして3つめは、コンピュータの能力が高くなったことです。
ハイスループット・スクリーニングを行うには設備と人手と時間が必要で、化合物を購入したり合成したりするための費用もかかりますが、バーチャル・スクリーニングなら、低コストですばやいスクリーニングが可能です。また、ハイスループット・スクリーニングでは、実際に集めた化合物しか調べることができない(せいぜい数万種)のに対し、バーチャル・スクリーニングなら、データベースにある化合物はすべて調べられます(数百万~数千万種)。しかも、現実には存在しない化合物であっても、コンピュータのなかに形をつくりさえすれば、調べることができるので、薬の候補の範囲が広がります。
さらに、原因分子の構造がわかっていない場合も、似た分子の構造がわかっていれば、その構造をもとに推定することができます。これは、ホモロジー・モデリングとよばれ、コンピュータの中だからこそ、できる技です。
計算で候補化合物をブラッシュアップ
原因分子に結合する化合物(ヒット化合物)が見つかっても、それがすぐに薬になるわけではありません。少量で効き目を示すものでなければ薬にはなりませんから、薬効が高いことが求められます。そこで、ヒット化合物の形を少しずつ変えながら、より薬効の高いもの(つまり、結合が強いもの)を探すことが必要になります。これをリード化合物の最適化(上の図の③)といいます。
この段階も、実験による方法と、計算による方法があります。実験では、ヒット化合物の構造を少し変えたものを実際に合成しては、それがより強い効果をもつかを調べます。細胞や動物を使うこともあります。
計算でも、ヒット化合物の構造を少しずつ変えることは同じですが、ドッキングよりも精密な計算方法が必要になります。
ドッキング法では、鍵穴となる原因分子の構造は動かさず固定されているが、そこに入る化合物は変形可能だとして、計算を行います。しかし、実際の体の中では、原因分子の構造はつねにゆらゆらと動いています。しかも、化合物がやってくると、構造を変えてから結合することもよくあります。ドッキング法では、こうした効果を無視しているので、結合の強さを正確に見積もることが難しいのです。
そこで、分子動力学(MD)シミュレーションと呼ばれる方法が使われます。これは、分子をつくっている原子を1つずつ、ニュートンの運動方程式に基づいて動かしていくものです。1983年にアメリカのカープラスらが、生体分子を扱うのに適した分子動力学法のソフトウェアCHARMMを開発して以降、さまざまなソフトが開発され、原因分子と化合物の結合を精密に計算することが可能になりました。
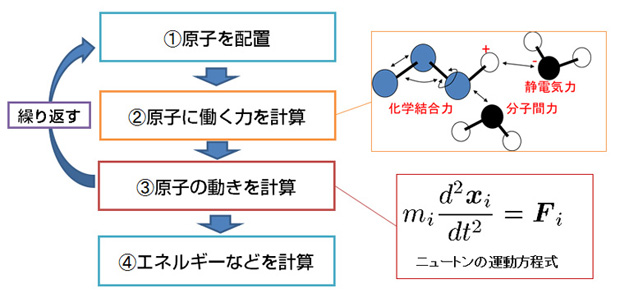
分子動力学シミュレーションの流れ
しかし、分子動力学法では、原因分子をつくっている何万もの原子を1個ずつ動かしていくので非常に計算量が多くなってしまいます。このため、普通のコンピュータで実行する場合には、原子がいくつか集まったグループを、1つの原子のように扱って計算するなどの工夫をして計算量を減らす必要があります。また、体の中では、原因分子は水に取り囲まれているのですが、水分子も計算するとなると計算量が増えてしまうので、水分子を採り入れることが難しいという問題もあります。
最先端の成果だけでなく、実用的な成果もあげた「京」
「京」では、原因分子と化合物に加え、周りの水分子も含むすべての原子についての分子動力学計算を行うことができました。5種類のタンパク質(病気の原因分子)に、それぞれ10種類程度の化合物が結合するようすを計算し、いちばん強く結合するものを予測したところ、正答率は50%以上になりました。一方、従来のドッキング法では、正答率は5%程度でした。この結果は、計算能力の高いスパコンで、ふんだんに計算を行えば、分子動力学法で最適なリード化合物を予測できることを示しています。つまり、分子動力学法による最適化が、将来は製薬の現場でも使えるということが実証されたのです。
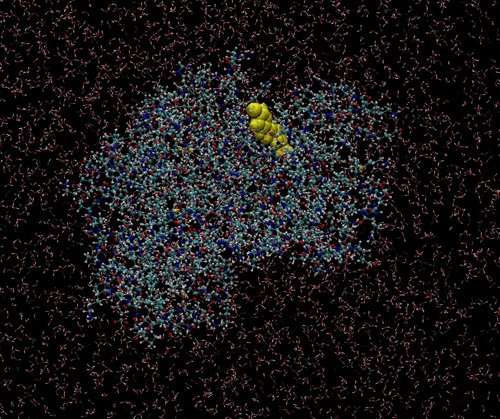
「京」で行われた分子動力学シミュレーションの例(画像提供:京都大学奥野恭史教授)
CDK2という酵素(がんの原因分子の1つ)に化合物(黄色)が結合するようすを計算した(本来は動画)。酵素の周りに広がっているのは水分子。
「京」ではもっと実用的な成果もあがっています。「京」の運用が始まったころ、「京」を利用した薬の設計を製薬業界全体で推進していくため、20社以上の製薬企業が参加するコンソーシアムが結成されました。このコンソーシアムでは、631種類のタンパク質と3000万種類の化合物の間でバーチャル・スクリーニングを行い(ペアの数は189億通り以上)、その計算結果をメンバー企業に配布しました。
このバーチャル・スクリーニングは、ドッキング法ではなく、デジカメで顔を自動認識するときに使われるパターン認識技術を用いており、「京」での計算時間は、8万ノードをすべて使ったとして換算すると5時間45分でした。タンパク質の種類は、メンバー企業の希望を聞いた上で選ばれているため、薬の設計のための基礎データとして、おおいに活用されそうです。
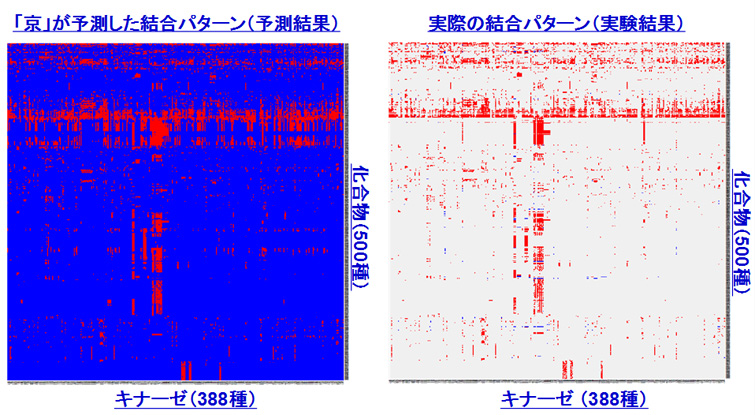
「京」で行ったバーチャル・スクリーニングの結果の一部(画像提供:京都大学奥野恭史教授)
キナーゼというタンパク質(酵素の一種)388種と500種の化合物の結合を調べた結果。赤いところは結合することを示す。右は、実際に実験で得られた結果をプロットしたもの。左の予測は、実験結果とよく一致している。予測では、実験が行われていない組み合わせについても結合するかどうかがわかる。
これまでにない薬の開発をめざすポスト「京」
ポスト「京」では、より「長時間」の分子動力学シミュレーションを「大規模」に行うことをめざします(重点課題(1)のサブ課題C 創薬ビッグデータ統合システムの開発)。「京」でも、一部の製薬企業が独自の分子動力学シミュレーションを行っていますが、ポスト「京」では、製薬企業が使いやすいようにアプリケーションとインターフェースを整え、より多くの製薬企業に使ってもらう計画です。
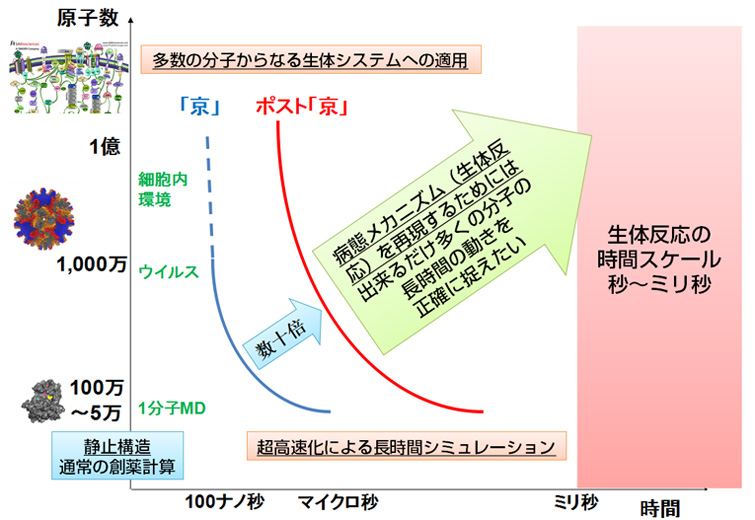
ポスト「京」がめざす分子動力学シミュレーション(画像提供:京都大学奥野恭史教授)
これまでより大きな原因分子も対象として、長時間のシミュレーションをめざす。
「長時間」とは、原子を動かす時間を長くするということです。分子動力学法では、原子を1フェムト秒(フェムトは10の-15乗)程度の時間刻みで少しずつ動かします。ですから「京」を使っても、シミュレーションできる時間の長さは10マイクロ秒(マイクロは10の-6乗)程度です(それでも100億回の計算が必要です)。しかし、結合の強さをほんとうに正確に計算するには、この100倍にあたる1ミリ秒の計算が必要です。原因分子に化合物が近づき、結合したり離れたりするのを繰り返す過程には、1ミリ秒程度かかるからです。
ポスト「京」では、1つ1つの組み合わせについて、1ミリ秒の分子動力学シミュレーションを行うのが目標です。実現すれば、これまでは、原因分子と化合物の結合場面だけを取り出した「予告編」しか見られなかったものが、最初から最後までを通した一編の映画として見られるようになるのです。これによって、どんな世界が開けてくるのか、研究者たちは楽しみにしています。
そして、「大規模」とは、調べられる組み合わせの数を増やすということです。「京」では、1マイクロ秒の分子動力学シミュレーションを、1週間で数百通り行うことが可能ですが、ポスト京では、1週間で数万通りの計算ができるようになるはずです。そうなれば、化合物が、別の分子に結合して副作用を起こす可能性や、想定していなかった原因分子に結合して別の薬効が見つかることもあるでしょう。ポスト「京」での分子動力学シミュレーションは、製薬企業にとって、実際の薬の設計に使える強力な武器となる可能性を秘めているのです。
その一方、製薬企業が取り組むには難しい、新しいタイプの薬の設計にも、ポスト「京」は取り組みます。例えば、最近、あるタンパク質と別のタンパク質が表面どうしでくっつくことが、病気の原因である場合が見つかってきています。この場合、従来の「鍵と鍵穴」のモデルでは、薬を設計することができません。そこで、タンパク質どうしの結合のようすをまず長時間の分子動力学シミュレーションで明らかにし、それを手がかりにして結合を邪魔する化合物の設計に進むことが考えられています。
薬の開発は、計算だけで行えるわけではなく、たくさんの要素技術が数珠つなぎになって10年以上の歳月をかけて行われます。開発のスピードを上げるには、1つ1つの要素技術を研ぎ澄まして最先端の技術をとり入れる必要があります。ポスト「京」は、その1つとして、今後、ますます重要性を増していくことでしょう。